Metaproteomics characterizes proteins expressed by microorganism communities (microbiome) present in environmental samples or a host organism. Mass spectrometry (MS)-based metaproteomics has catalyzed new discoveries into the functional dynamics of microbiomes (Wilmes et al 2015, doi: 10.1002/pmic.201500183). Metaproteomic informatics is distinctly challenging due to the large databases and complex processing steps involved. This challenge limits widespread use of metaproteomics. Through modular workflows, we demonstrate the use of the Galaxy bioinformatics framework as a metaproteomic informatics solution. These workflows enable new discoveries from diverse communities such as dental plaques, bronchoalveolar lavage fluid (BALF), lung tissue, and cervical-vaginal fluid (CVF). Our results demonstrate the power of discovery metaproteomics to add functional understanding to microbiomes, beyond what is possible using traditional metagenomic approaches.
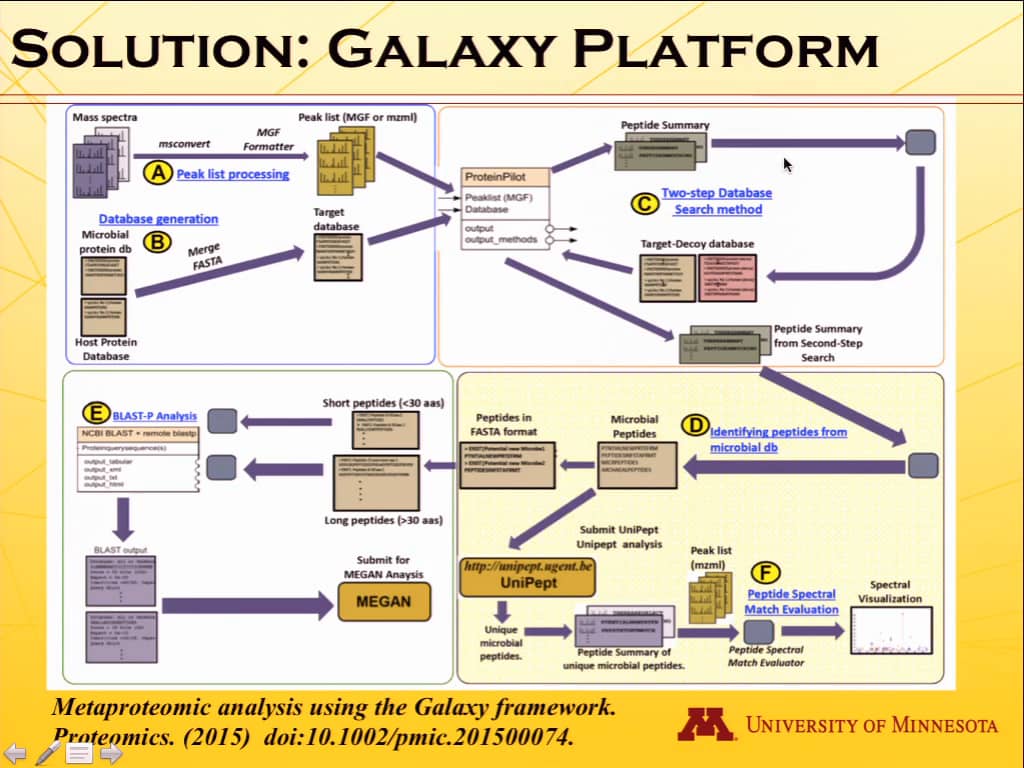
Several types of human biospecimens, were subjected to trypsin digestion, C18 column purification, offline fractionation by high-pH reverse phase chromatography, and mass spectrometry (MS) using the Orbitrap Velos instrument with Orbitrap MS and Higher energy collision dissociation MS/MS mode. In a modular workflow, RAW files converted to MGF format using msconvert were searched against custom databases using ProteinPilot with the two-step method (z.umn.edu/metaproteomics1). Subsequent automated workflows also enable text processing, BLAST-P analysis and visualization of peptide sequence match (PSM) quality (Jagtap et al 2015; doi: 10.1002/pmic.201500074). The workflow output results are compatible with tools for taxonomic and functional characterization (Unipept and MEGAN5). MEGAN5 was used to generate functional characterization of the metaproteome using SEED and KEGG pathway analysis.
To address the challenges in metaproteomics analysis, we used the Galaxy workflows on microbiomes.
For example, to study sucrose-induced dysbiosis in taxonomically diverse oral microcosms, plaque samples from each of twelve children at high risk for caries were grown in paired biofilm reactors, in the presence and absence of sucrose pulsing. Functional analysis of generated metaproteomic data indicated that sucrose-induced changes in protein relative abundance patterns for several metabolic pathways, and those patterns were conserved among taxonomically diverse samples (Rudney et al 2015, doi: 10.1186/s40168-015-0136-z).